pc端手机网站 viewport 自适应百度竞价推广账户
检索到目标数据集后,开始数据挖掘,本文以阿尔兹海默症数据集GSE1297为例
目录
离群值处理
删除 低表达基因
函数归一化,矫正差异
数据标准化—log2处理
完整代码
上节围绕着探针ID和基因名称做了一些清洗工作,还做了重复值检查,空值删除操作。
#查看重复值
table(duplicated(matrix$Gene.Symbol))#去掉缺失值
matrix_na = na.omit(matrix) #基因名称为空删除
matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]离群值处理
用于处理异常值,将超出一定范围的值替换为中位数,以减少异常值对后续分析的影响。
#数据离群处理
#处理极端值
#定义向量极端值处理函数
#用于处理异常值,将超出一定范围的值替换为中位数,以减少异常值对后续分析的影响。
dljdz=function(x) {DOWNB=quantile(x,0.25)-1.5*(quantile(x,0.75)-quantile(x,0.25))UPB=quantile(x,0.75)+1.5*(quantile(x,0.75)-quantile(x,0.25))x[which(x<DOWNB)]=quantile(x,0.5)x[which(x>UPB)]=quantile(x,0.5)return(x)
}#第一列设置为行名
matrix_leave=matrix_finalboxplot(matrix_leave,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave)#处理离群值
matrix_leave_res=apply(matrix_leave,2,dljdz)boxplot(matrix_leave_res,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave_res)
删除 低表达基因
方案1 :仅去除在所有样本里表达量都为零的基因(或者平均值小于1)
方案2:仅保留在一半(50%,75%...自己选择)以上样本里表达的基因
#删除 低表达基因#仅去除在所有样本里表达量都为零的基因(平均值小于1)# 计算基因表达矩阵的平均值
gene_avg <- apply(matrix_final, 1, mean)
# 根据阈值筛选低表达基因
filtered_genes_1 <- matrix_final[gene_avg >= 1, ] # 表达量平均值小于1的过滤
dim(filtered_genes_1)#+================================
#常用过滤标准2(推荐):
#仅保留在一半以上样本里表达的基因# 计算基因表达矩阵每个基因的平均值
gene_means <- rowMeans(matrix_final)# 计算基因平均值的排序百分位数
gene_percentiles <- rank(gene_means) / length(gene_means)# 获取阈值
threshold <- 0.25 # 删除后25%的阈值
#threshold <- 0.5 # 删除后50%的阈值
# 根据阈值筛选低表达基因
filtered_genes_2 <- matrix_final[gene_percentiles > threshold, ]# 打印筛选后的基因表达矩阵
dim(filtered_genes_2)boxplot(filtered_genes_2,outline=FALSE, notch=T, las=2) ##出箱线图
#dim(filtered_genes)#+删除 低表达基因 得到filtered_genes_1 删除平均表达小于1的 得到filtered_genes_2 删除后25%的函数归一化,矫正差异
#1.归一化不是绝对必要的,但是推荐进行归一化。
#有重复的样本中,应该不具备生物学意义的外部因素会影响单个样品的表达,
#例如中第一批制备的样品会总体上表达高于第二批制备的样品,假设所有样品表达值的范围和分布都应当相似,
#需要进行归一化来确保整个实验中每个样本的表达分布都相似。
#2.归一化要在log2标准化之前做
library(limma) exprSet=normalizeBetweenArrays(filtered_genes_2)boxplot(exprSet,outline=FALSE, notch=T, las=2) ##出箱线图## 这步把矩阵转换为数据框很重要
class(exprSet) ##注释:此时数据的格式是矩阵(Matrix)
exprSet <- as.data.frame(exprSet)数据标准化—log2处理
如果表达量的数值在50以内,通常是经过log2转化后的。如果数字在几百几千,则是未经转化的。
GSE数据集的注释部分会有说明,常见的标准化处理方法有3种:RMA算法、GC-RMA算法、MAS5算法
#标准化 表达矩阵自动log2化
qx <- as.numeric(quantile(exprSet, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||(qx[6]-qx[1] > 50 && qx[2] > 0) ||(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)## 开始判断
if (LogC) { exprSet [which(exprSet <= 0)] <- NaN## 取log2exprSet_clean <- log2(exprSet)print("log2 transform finished")
}else{print("log2 transform not needed")
}筛选后的基因表达矩阵的箱线图
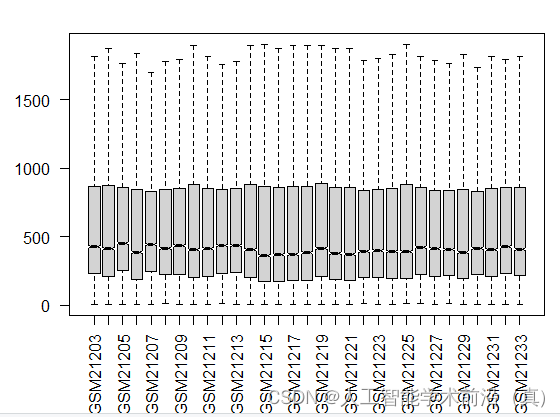
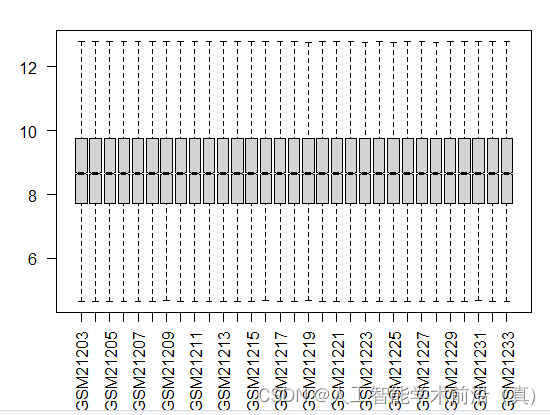
经过数据清洗后的箱线图
完整代码
# 安装并加载GEOquery包
library(GEOquery)# 指定GEO数据集的ID
gse_id <- "GSE1297"# 使用getGEO函数获取数据集的基础信息
gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!# 提取基因表达矩阵
expression_data <- exprs(gse_info[[1]])# 提取注释信息
#annotation <- featureData(gse_info[[1]])#查看平台文件列名
colnames(annotation)#打印项目文件列表
dir() # 读取芯片平台文件txt
platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")#查看平台文件列名
colnames(platform_file)# 假设芯片平台文件中有两列,一列是探针ID,一列是基因名
#probe_names <- platform_file$ID
#gene_symbols <- platform_file$Gene.Symbol
platform_file_set=platform_file[,c(1,11)]#一个探针对应多个基因名,保留第一个基因名
ids = platform_file_set
library(tidyverse)
test_function <- apply(ids,1,function(x){paste(x[1],str_split(x[2],'///',simplify=T),sep = "...")})
x = tibble(unlist(test_function))colnames(x) <- "ttt"
ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
dim(ids)#将Matrix格式表达矩阵转换为data.frame格式
exprSet <- data.frame(expression_data)
dim(exprSet)#给表达矩阵新增加一列ID
exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列
dim(exprSet)
#矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并
express <- merge(x = exprSet, y = ids, by.x = "ID")#删除探针ID列
express$ID =NULLdim(express) matrix = express
dim(matrix)
#查看多少个基因重复了
table(duplicated(matrix$Gene.Symbol))#把重复的Symbol取平均值
matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOLdim(matrix)matrix_na = na.omit(matrix) #去掉缺失值
dim(matrix_na)matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]
dim(matrix_final)matrix_final <- subset(matrix_final, select = -1) #删除Symbol列(一般是第一列)
dim(matrix_final)
#+ 经过注释、探针名基因名处理、删除基因名为空值、删除缺失值 得到最终 matrix_final
#+==================================================================================
#+========================================================================================#+==================================================================================
#+========================================================================================
#+增加#(2)离群值处理#数据离群处理
#处理极端值
#定义向量极端值处理函数
#用于处理异常值,将超出一定范围的值替换为中位数,以减少异常值对后续分析的影响。
dljdz=function(x) {DOWNB=quantile(x,0.25)-1.5*(quantile(x,0.75)-quantile(x,0.25))UPB=quantile(x,0.75)+1.5*(quantile(x,0.75)-quantile(x,0.25))x[which(x<DOWNB)]=quantile(x,0.5)x[which(x>UPB)]=quantile(x,0.5)return(x)
}#第一列设置为行名
matrix_leave=matrix_finalboxplot(matrix_leave,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave)#处理离群值
matrix_leave_res=apply(matrix_leave,2,dljdz)boxplot(matrix_leave_res,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave_res)#+增加#(2)离群值处理
#+==================================================================================
#+========================================================================================#+==================================================================================
#+========================================================================================
#删除 低表达基因#仅去除在所有样本里表达量都为零的基因(平均值小于1)# 计算基因表达矩阵的平均值
gene_avg <- apply(matrix_final, 1, mean)
# 根据阈值筛选低表达基因
filtered_genes_1 <- matrix_final[gene_avg >= 1, ] # 表达量平均值小于1的过滤
dim(filtered_genes_1)#+================================
#常用过滤标准2(推荐):
#仅保留在一半以上样本里表达的基因# 计算基因表达矩阵每个基因的平均值
gene_means <- rowMeans(matrix_final)# 计算基因平均值的排序百分位数
gene_percentiles <- rank(gene_means) / length(gene_means)# 获取阈值
threshold <- 0.25 # 删除后25%的阈值
#threshold <- 0.5 # 删除后50%的阈值
# 根据阈值筛选低表达基因
filtered_genes_2 <- matrix_final[gene_percentiles > threshold, ]# 打印筛选后的基因表达矩阵
dim(filtered_genes_2)boxplot(filtered_genes_2,outline=FALSE, notch=T, las=2) ##出箱线图
#dim(filtered_genes)#+删除 低表达基因 得到filtered_genes_1 删除平均表达小于1的 得到filtered_genes_2 删除后25%的
#+==================================================================================
#+========================================================================================#+==================================================================================
#+========================================================================================
### limma的函数归一化,矫正差异 ,表达矩阵自动log2化#1.归一化不是绝对必要的,但是推荐进行归一化。
#有重复的样本中,应该不具备生物学意义的外部因素会影响单个样品的表达,
#例如中第一批制备的样品会总体上表达高于第二批制备的样品,假设所有样品表达值的范围和分布都应当相似,
#需要进行归一化来确保整个实验中每个样本的表达分布都相似。
#2.归一化要在log2标准化之前做library(limma) exprSet=normalizeBetweenArrays(filtered_genes_2)boxplot(exprSet,outline=FALSE, notch=T, las=2) ##出箱线图## 这步把矩阵转换为数据框很重要
class(exprSet) ##注释:此时数据的格式是矩阵(Matrix)
exprSet <- as.data.frame(exprSet)#标准化 表达矩阵自动log2化
qx <- as.numeric(quantile(exprSet, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||(qx[6]-qx[1] > 50 && qx[2] > 0) ||(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)## 开始判断
if (LogC) { exprSet [which(exprSet <= 0)] <- NaN## 取log2exprSet_clean <- log2(exprSet)print("log2 transform finished")
}else{print("log2 transform not needed")
}boxplot(exprSet_clean,outline=FALSE, notch=T, las=2) ##出箱线图
### limma的函数归一化,矫正差异 ,表达矩阵自动log2化 得到exprSet_clean
#+==================================================================================
#+========================================================================================# saving all data to the path
save(exprSet_clean, file ="exprSet_clean_75percent_filter.RData")上述处理得到了干净的基因表达矩阵,数据部分已经没有问题,但是在做数据挖掘(差异分析、富集分析等)之前还有一项准备工作,要将数据样本进行分组,即患病组、对照组。